
Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Бяс при деца
Медицински експерт на статията

Последно прегледани: 04.07.2025
Бяс, или хидрофобия, е остро вирусно заболяване, предавано чрез ухапване от заразено животно, с увреждане на нервната система и развитие на тежък енцефалит с фатален изход.
Епидемиология
Вирусът на бяс, бич за общественото здраве от древни времена, понастоящем причинява приблизително 59 000 смъртни случая всяка година, почти всички от които се предават чрез ухапвания от кучета. Това има значително икономическо въздействие върху развиващите се страни, особено в Африка и Азия, които могат да понесат най-малко подобни загуби. Въпреки почти 100% смъртност, бясът при кучетата е напълно предотвратимо заболяване и историческите примери за неговото премахване в развития свят свидетелстват за това. [ 1 ]
Причини бяс
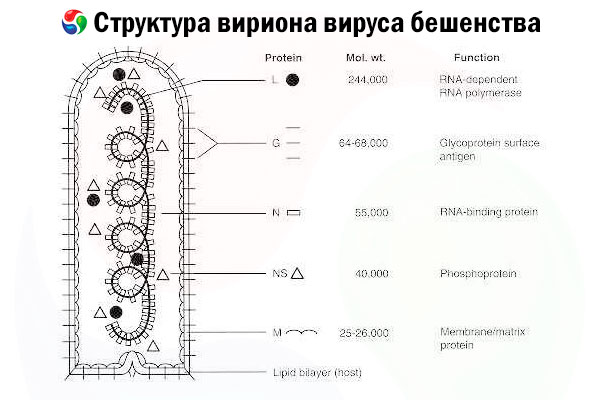
Причинителят е вирусът на бяс (RV), РНК вирус с отрицателна верига от семейство рабдовируси, с размери приблизително 60 nm × 180 nm.
Състои се от вътрешно протеиново ядро, или нуклеокапсид, съдържащо нуклеинова киселина, и външна мембрана, съдържащ липиди бислой, покрит с трансмембранни гликопротеинови шипове. Има относително проста модулна геномна структура и кодира пет структурни протеина:
- РНК-зависима РНК полимераза (L),
- нуклеопротеин (N),
- фосфорилиран протеин (P),
- матричен протеин (М) и
- гликопротеин на външната повърхност (G).
N, P и L протеините, заедно с геномната РНК, образуват рибонуклеопротеиновия комплекс. G е единственият антиген на ротавируса, способен да индуцира производството на неутрализиращи антитела, които са основните имунни ефектори срещу летална инфекция с ротавирус. От друга страна, рибонуклеопротеиновият комплекс е доказано, че е основният антиген на ротавируса, способен да индуцира CD4+ Т-клетки, което може да засили производството на неутрализиращи антитела, причинени от ротавируса, чрез интраструктурно разпознаване на антигена.[ 2 ] Рибонуклеопротеиновият комплекс може да играе важна роля в установяването на имунологична памет и дългосрочен имунитет.[ 3 ]
 [
[ Класификация и видове антигени
Родът Lyssavirus включва вируса на бяс и антигенно и генетично свързани вируси на бяс: вирусите на прилепите Lagos, Mokola и Duvenhage, както и два предполагаеми подтипа на европейските лисавируси на прилепите. Проучванията за кръстосана защита показват, че животните, имунизирани с традиционни ваксини срещу бяс, може да не са напълно защитени при заразяване с други лисавируси.
Вирусите на бяс могат да бъдат класифицирани като фиксирани (адаптирани чрез преминаване през животни или клетъчна култура) или улични (див тип). Използването на моноклонални антитела и генетично секвениране за диференциране на уличните вируси на бяс е помогнало за идентифициране на вирусни варианти, произхождащи от големи резервоари гостоприемници по целия свят, и за предполагане на вероятни източници на експозиция при хора, когато в случая на пациента липсва анамнеза за окончателно ухапване от животно.[ 8 ]
Патогенеза
Основен резервоар и източник на инфекция сред дивите животни са вълци, лисици, чакали, прилепи, а сред домашните животни - кучета и котки, рядко - коне, говеда, свине, плъхове и др. Предаването на инфекцията от човек на човек, макар и възможно, е изключително рядко. Това е типична зоонозна инфекция. Хората се заразяват с бяс предимно от кучета.
След ухапване на човек от болно животно, вирусът се размножава в мускулната тъкан на мястото на ухапването, а след това, достигайки краищата на сетивните периферни нерви, се разпространява центростремително, достигайки до моторните неврони. Времето, необходимо за придвижване на вируса и засягане на мозъка, зависи от мястото на ухапване. При тежки ухапвания по главата и лицето, вирусът може да достигне централната нервна система за 15-20 дни, а при незначителни увреждания на кожата на торса и крайниците и следователно малка доза от патогена, процесът на придвижване на вируса към централната нервна система може да се забави с няколко месеца или дори до 1-1,5 години. След като достигне централната нервна система, вирусът се фиксира в тъканите на главния и гръбначния мозък, главно в невроните на продълговатия мозък, рога на Амон и основата на мозъка. В гръбначния мозък най-засегнати са задните рога. От централната нервна система вирусът центробежно по нервните стволове достига до слюнчените жлези, където се размножава и се отделя със слюнка.
Концепции в патогенезата на бяс
Вирусът на бяс (RV) има широк кръг от гостоприемници и може да зарази почти всички бозайници. Въпреки че са съобщени няколко пътя на предаване на RV, естествената инфекция най-често се осъществява чрез ухапване. В допълнение към ухапванията, консумацията на трупове, заразени с RV, може да насърчи инфекцията с вируса на бяс при арктическите лисици, а контактът на RV с лигавиците е установен като друг възможен път на предаване.[ 9 ] При някои необичайни обстоятелства, като например случайно изпускане на RV като аерозол в лаборатория или RV като аерозол в пещери, обитавани от голям брой прилепи,[ 10 ] може да възникне аерозолно предаване.
Все още не е ясно дали уличните щамове на ротавирус (RV) и адаптираните към мишки или адаптирани към тъканни култури щамове на RV се репликират на мястото на инокулация, преди да влязат в ЦНС. Докато експерименталното интрамускулно инфектиране на млади хамстери или миещи мечки с уличен RV разкрива репликация на RV в клетки на набраздени мускули, преди вирусът да нахлуе в аксоните на моторните неврони през невромускулните връзки,[ 11 ],[ 12 ] интрамускулното инфектиране на мишки с адаптиран към мишки CVS-24 RV показва, че RV мигрира директно към ЦНС без предварителна репликация на мястото на инокулация.[ 13 ] Веднъж попаднал в терминалите на немиелинизираните аксони, RV се транспортира ретроградно до клетъчното тяло.
Последните открития показват, че транспортът на аксонални везикули може да представлява ключова стратегия за движение на вириони на дълги разстояния в аксони.[ 14 ] Изчислено е, че RV мигрира в аксоните със скорост от 3 mm/h.[ 15 ] След това инфекцията се разпространява през верига от неврони, свързани чрез синаптични връзки. Точният механизъм, който насърчава транссинаптичното разпространение, обаче все още е неизвестен. След заразяване на мозъка, вирусът се разпространява центробежно към периферната и автономната нервна система в много периферни органи.[ 16 ] В последния етап от инфекциозния цикъл RV мигрира към слюнчените жлези; след репликация в мукогенни ацинарни клетки, той се освобождава в слюнката и е готов за предаване на следващия гостоприемник.[ 17 ]
По отношение на патологията, индуцирана от вируса на бяс, апоптотичната клетъчна смърт е предложена като потенциален патогенен механизъм в експериментални модели на бяс при мишки, заразени с фиксиран щам на RV.[ 18 ] Патогенен механизъм, който може да допринесе за дълбоката дисфункция на ЦНС, характерна за бяс, може да бъде нарушена невронна функция. Доказано е, че генната експресия е значително намалена в заразените с RV неврони, което води до общо потискане на протеиновия синтез,[ 19 ] и няколко проучвания показват нарушена невротрансмисия след RV инфекция. Jiang демонстрира, че свързването на антагонист на ацетилхолиновия рецептор към заразени хомогенати от мозъка на плъхове е намалено в сравнение с контролите.[ 20 ] Нарушено освобождаване и свързване на серотонин, невротрансмитер, участващ в контрола на цикъла на съня, възприятието за болка и поведението, също са наблюдавани в мозъка на заразени с RV плъхове.[ 21 ], [ 22 ] В допълнение към повлияването на невротрансмисията, инфекцията на дясната камера може да засегне и йонните канали. Заразените клетки на миши невробластом показват намалена функционална експресия на волтажно-зависими натриеви канали, което може да предотврати акционните потенциали и в крайна сметка да доведе до функционално увреждане. [ 23 ]
В допълнение към липсата на сериозни патологични лезии в ЦНС, повечето случаи на бяс при хора не предизвикват имунен отговор 7 до 10 дни след началото на клиничните признаци. Тези дълбоки разлики между патогенезата на бяса и тази на повечето други вирусни или бактериални инфекции на ЦНС се подкрепят допълнително от факта, че имуносупресията е или неефективна, или вредна за изхода на бяса.[ 24 ] Ниското ниво на имунен отговор, често наблюдавано при жертви на бяс, е озадачаващо, защото не може да се обясни с лошата имуногенност на антигените на ротавируса. Всъщност, ротавирусният G и нуклеокапсидният протеин са мощни B- и T-клетъчни антигени, когато се прилагат парентерално.[ 25 ] Възможно обяснение за ниската степен на имунен отговор срещу ротавируса при хора или животни с бяс може да бъде, че ротавирусната инфекция на ЦНС причинява имуносупресия,[ 26 ] и е предложено, че ротавирусът използва подривна стратегия, включваща предотвратяване на апоптоза и унищожаване на нахлуващите Т-клетки.[ 27 ]
Атенюираните щамове на ротавирус, адаптирани към неневронни клетки, се различават значително от патогенните щамове на уличния ротавирус по своята невроинвазивност, която се отнася до способността им да нахлуват в ЦНС от периферни места. В тази връзка, адаптираните към тъканни култури щамове на ротавирус или нямат, или имат само ограничена способност да нахлуват в ЦНС от периферни места, докато щамовете на уличния ротавирус или адаптираните към мишки щамове на ротавирус, като CVS-24, са силно инвазивни.[ 28 ] Ключови фактори, участващи в невроинвазията на ротавируса, включват вирусно усвояване, аксонен транспорт, транссинаптично разпространение и скорост на вирусна репликация.
Доскоро познанията ни за патогенезата на ротавирусната инфекция (РВ) бяха ограничени и се основаваха предимно на описателни изследвания на улични щамове на РВ или експериментални инфекции с атенюирани щамове, адаптирани в лаборатория. Появата на технологията за обратна генетика ни позволи да идентифицираме вирусните елементи, които определят патогенния фенотип на РВ, и да разберем по-добре механизмите, участващи в патогенезата на бяс.
Идентифициране на вирусни елементи, контролиращи придобиването, разпространението и репликацията на вируса на бяс
- Вирусни елементи, участващи в улавянето на вируса
Инфекцията с ротавирусен вирус (RV) започва с прикрепване на вируса към предполагаем клетъчен рецептор. Въпреки че няколко мембранни повърхностни молекули са предложени като RV рецептори, включително никотиновия ацетилхолинов рецептор,[ 29 ] молекулата на невронната клетъчна адхезия[ 30 ] и нискоафинитетния невротрофинов рецептор p75 NTR,[ 31 ] все още не е ясно дали тези молекули действително играят роля в жизнения цикъл на вируса на бяс. В този контекст наскоро беше показано, че взаимодействието RV G–p75 NTR не е необходимо за RV инфекция на първични неврони.[ 32 ] След свързване с рецептора, RV се интернализира чрез адсорбционна или рецептор-медиирана ендоцитоза.[ 33 ] Ниското pH на средата в ендозомалния компартмент след това индуцира конформационни промени в RV G, които задействат сливането на вирусната мембрана с ендозомната мембрана, като по този начин освобождават RNP в цитоплазмата.[ 34 ] При вирусите RV G играе критична роля в вирусното усвояване, най-вероятно чрез взаимодействия с предполагаеми клетъчни рецептори, които улесняват бързото усвояване. В тази връзка е доказано, че патогенността на адаптирани към тъканни култури щамове на RV (напр. ERA, HEP и CVS-11) корелира с наличието на детерминанта, разположена в антигенен сайт III на G протеина. [ 35 ] Мутация Arg → Gln в позиция 333 в този антигенен сайт на ERA G протеина води до седемкратно забавяне на интернализацията на варианта Gln333 RV в сравнение с варианта от див тип. Мутацията Asn194→Lys194 в RV G, която обяснява повторната поява на патогенния фенотип, е свързана със значително намаляване на времето за интернализация.[ 36 ] Освен това, експерименти с химерни RV показват, че времето, необходимо за интернализация на RV вириони, е значително увеличено и патогенността е силно намалена след заместване на G гена на силно патогенния SB RV щам, който е получен от кДНК клон на получения от сребро щам RV-18, свързан с прилепи,[ 37 ] с този на силно атенюирания SN щам, който е изолиран от кДНК клон на ваксиналния щам SAD B19 RV.[ 38 ] Взети заедно, тези данни подкрепят идеята, че кинетиката на усвояване на вируса, която е функция на RV G, е основен фактор за патогенността на RV.
- Вирусни елементи, участващи в разпространението и предаването на вируси
Уникално свойство на вируса на бяс е способността му да се разпространява от клетка на клетка. Наблюдението, че вариантът Gln333 ERA губи pH-зависима активност за клетъчно-клетъчно сливане in vitro [ 39 ] и показва значително намалена способност за разпространение от клетка на клетка [ 40 ], предполага, че RV G също играе ключова роля в разпространението от клетка на клетка и следователно в предаването на вируса, вероятно чрез своята фузиогенна активност. Тази възможност се подкрепя допълнително от констатацията, че скоростта на разпространение на патогенния RV ревертантен SPBNGAK е почти два пъти по-висока от тази, определена за непатогенния вариант SPBNGA. Интересно е, че мутацията Asn 194 → Lys 194 в G SPBNGAK причинява изместване на прага на pH за мембранно сливане към по-високо pH, което подкрепя хипотезата, че по-високият праг на pH за мембранно сливане е свързан с повишено разпространение на вируса. [ 41 ]
Проучвания на трансневронални индикатори за RV инфекция при плъхове [ 42 ] и резус маймуни [ 43 ] показват, че вирусът на бяс мигрира изключително в ретроградна посока в аксоните. Въпреки че няколко RV протеина участват в невронните транспортни механизми, RV G изглежда играе преобладаваща роля в трансневроналното разпространение на RV инфекцията. Например, докато периферната инфекция с вирус на инфекциозна анемия по конете (EIAV), псевдотипизиран с RV G, води до вирусен трансфер към гръбначния мозък, същият EIAV, псевдотипизиран с вирус на везикуларен стоматит G, не успява да навлезе в нервната система. [ 44 ] Освен това, вирусното разпространение на мутанта ERA G Arg 333 → Gln 333 в ЦНС е установено, че е силно намалено в сравнение с мутанта от див тип, което допълнително предполага функция на интактния RV G в транссинаптичното разпространение. Най-убедителните доказателства за важна роля на RV G в транссинаптичния транспорт обаче идват от вътречерепна инфекция на мишки с рекомбинантен G-дефицитен RV вирус, което показва, че инфекцията остава ограничена до неврони на мястото на инокулация, без никакви доказателства за разпространение към вторични неврони.[ 45 ] Вероятно е обаче, освен RV G, RV M също да играе роля в разпространението на вируса и следователно в транссинаптичния транспорт. В тази връзка беше показано, че разпространението на химерния вариант SN-BMBG RV, който съдържа както M, така и G от силно патогенния SB, е значително по-високо от разпространението на химерния вариант SN-BG или SN-BM, които съдържат съответно G и M от SB, което предполага, че оптималното взаимодействие на M с G може да играе важна роля в разпространението на вируса от клетка на клетка.[ 46 ] Тъй като RV M поддържа пъпкуването на вируса,[ 47 ] вероятно е по-ефективното разпространение на химерния вариант RV SN-BMBG да се дължи на оптималното пъпкуване на вируса на постсинаптичната мембрана.
Последните проучвания показват, че взаимодействието между RV P и леката верига на динеина свързва RV RNP с транспортната система на клетката гостоприемник, като по този начин улеснява ретроградния аксонен транспорт на вируса.[ 48 ],[ 49 ] Въпреки това, периферна инфекция на възрастни мишки показва, че делецията на LC8 свързващия домейн на RV P не предотвратява навлизането на вируса в ЦНС, което предполага, че RV протеинът не участва пряко в ретроградното аксонално разпространение на RV.[ 50 ]
- Вирусни елементи, които контролират вирусната репликация
За разлика от много други вируси, като например грипния вирус, патогенността на RV е обратно пропорционална на скоростта на синтез на вирусна РНК и производството на инфекциозни вирусни частици. Сравнението на нивата на вирусна мРНК и геномна РНК, произвеждани от различни химерни вируси, предполага, че транскрипцията и репликацията на вирусна РНК се регулират от множество фактори, включително RV M, който е идентифициран като транс-действащ фактор, който медиира прехода от начални високи нива на синтез на мРНК към синтез на геномна РНК.[ 51 ] Освен това, M от всички рабдовируси е способен да изключи експресията на вирусни гени чрез свързване с RNP, което води до образуването на силно кондензирана структура, подобна на гръбнак, която не е в състояние да поддържа синтеза на РНК.
За да се идентифицират други вирусни елементи, които контролират патогенността чрез регулиране на вирусната репликация, 5'-терминалните последователности на силно патогенния SB щам бяха заменени поетапно с последователности от силно атенюирания SN ваксинален щам, което доведе до рекомбинантни вируси SB2 (терминална последователност [TS] + L), SB3 (TS + L + псевдоген [Ψ]), SB4 (TS + L + Ψ + G) и SB5 (TS + L + Ψ + G + M). Интрамускулната инфекция с родителските SB и SN вируси и химерните RV SB2, SB3, SB4 и SB5 предизвика най-високи нива на смъртност при SB-инфектирани мишки и никаква заболеваемост или смъртност при SN-инфектирани мишки. Заместването на TS, L и SB със съответните елементи от SN доведе до умерено намаляване на заболеваемостта и смъртността, а допълнителен обмен на G или G плюс M силно намали или напълно премахна вирусната патогенност.
Фенотипната характеристика на тези див тип и химерни ротавируси (RV) в тъканна култура разкри, че патогенността на даден RV е обратнопропорционална на способността му да се репликира в невронните клетки. Въпреки че SB се репликира на нива почти 1000 пъти по-ниски от SN, и заместването на TS, L и в SB с нива на SN има малък ефект върху кинетиката на вирусния растеж, допълнителното заместване на G или G плюс M на SB със съответните SN гени води до 1-log увеличение на вирусното производство, което предполага, че кинетиката на репликация на вирусна РНК, както и производството на вирусни частици, се контролират до голяма степен от RV G протеина. Това заключение се подкрепя от данни, получени с RV G варианти, които се различават по една аминокиселина в техните G протеини. Патогенният вариант на вируса на бяс SPBNGAK 194 е продуцирал вирусен титър в NA клетки, който е бил с 1 log по-нисък от този, произведен от непатогенния вариант SPBNGAN 194, а PCR анализът в реално време показа, че скоростите на транскрипция и репликация на вирусната РНК в SPBNGAK-инфектирани NA клетки са 5 и 10 пъти по-високи, отколкото в SPBNGAK-инфектирани NA клетки.[ 52 ] Допълнителни доказателства за обратна корелация между патогенността и скоростта на синтез на вирусна РНК и производството на вирусни частици са предоставени от мишки, инфектирани с химерни рекомбинантни вируси, в които G и M гените на атенюирания SN щам са заменени с тези на силно патогенния SB щам. Тези експерименти разкриват значително увеличение на патогенността на родителския SN щам, носещ RV G, в сравнение с патогенния SB щам. Патогенността се увеличава допълнително, когато както G, така и M от SB са въведени в SN.
Заместването на G или M или и на двата в SN със съответните гени от SB е свързано със значително намаляване на скоростта на производство на вирусни частици, както и на скоростта на синтез на вирусна РНК. Тези данни показват, че както G, така и M играят важна роля в патогенезата на RV чрез регулиране на вирусната репликация. Констатацията, че заместването на G или G плюс M в SN с G или G плюс M от SB води до умерено до силно намаляване на транскрипцията и репликацията на вирусната РНК, съответно, докато заместването само на M в SN с M от SB води до силно увеличение на транскрипцията и репликацията на вирусната РНК, показва, че RV G също има важна регулаторна функция в транскрипцията/репликацията на вирусната РНК, самостоятелно или чрез взаимодействие с M протеина. Механизмът, чрез който RV G генът контролира синтеза на вирусна РНК, е неизвестен. Някои нуклеотидни последователности в RV G гените, като тези, включващи кодоните за Arg333 и Lys 194, са идентифицирани като мишени за клетъчни miRNA. Показано е, че разпознаването на мишени от клетъчни miRNA може да доведе до положителна или отрицателна регулация на вирусната репликация. [ 53 ] Заместванията Arg 333 → Glu 333 или Lys 194 → Ser 194 в генната последователност на RV G водят до премахване на miRNA таргетните последователности, което от своя страна е свързано със значително увеличение на скоростта на синтез на вирусна РНК [Faber M, Thomas Jefferson University, PA, USA, Unpubliced Data], което предполага, че miRNA в клетките на гостоприемника също играят важна роля в регулирането на репликацията на RV, както е показано за други РНК вируси, включително вируса на везикуларен стоматит и HCV. [ 54 ], [ 55 ]
Регулирането на вирусната репликация изглежда е един от важните механизми, участващи в патогенезата на ротавирусната инфекция (РВ). За да избегнат имунния отговор и да запазят целостта на невронната мрежа, патогенните щамове на РВ, но не и атенюираните щамове, могат да регулират скоростта си на растеж. По-ниската скорост на репликация вероятно е от полза за патогенните щамове на РВ, като запазва невронната структура, която тези вируси използват, за да достигнат до ЦНС. Друго обяснение за по-ниската скорост на репликация на патогенния РВ е, че за да избегне ранното откриване от имунната система на гостоприемника, вирусът поддържа минимални нива на експресия на своите антигени.
Връзка между експресията на RV G, апоптозата и патогенността
Добре известно е, че щамовете на вируса на уличната бяс, които са значително по-патогенни от щамовете, адаптирани към тъканни култури, експресират много ограничени нива на G и не индуцират апоптоза до късен етап от инфекциозния цикъл, което предполага, че патогенността на даден вирусен щам е обратнопропорционална на експресията на RV G и способността му да индуцира апоптоза.[ 56 ] Директни доказателства за корелация между нивото на експресия на G и степента на апоптоза са получени с рекомбинантния RV SPBNGA-GA, който носи два идентични G гена и свръхекспресира RV G.[ 57 ] Морфологични изследвания на невронни култури, инфектирани с този рекомбинантен RV, показват, че клетъчната смърт е значително повишена паралелно със свръхекспресията на RV G и че апоптозата е основният механизъм, участващ в смъртта, медиирана от RV G. По-специално, намаляването на F-актин оцветяването след SPBNGA-GA инфекция е в съответствие с индуцираната от апоптоза деполимеризация на актинови филаменти. Освен това, броят на TUNEL-позитивните ядра в SPBNGA-GA-инфектираните неврони е значително увеличен в сравнение с този в неинфектираните и SPBNGA-инфектираните неврони. Въпреки това, механизмът, чрез който генът RV G медиира процеса на апоптотична сигнализация, остава до голяма степен неизвестен. Предполага се, че експресията на RV G над определен праг сериозно нарушава клетъчната мембрана. Много е вероятно апоптотичните клетки да не се изчистват бързо в ЦНС и следователно да претърпят вторична некроза. [ 58 ] От друга страна, RV инфекцията и по-специално свръхекспресията на RV G протеина може да доведе до пироптоза, път на клетъчна смърт, подобен на апоптозата, който, за разлика от апоптозата, включва активиране на каспаза 1 и по този начин води до некроза. [ 59 ] Степента на некроза или пироптоза, индуцирана от RV инфекция, вероятно играе критична роля в индуцирането на антивирусен имунитет. Докато апоптотичните клетки поддържат целостта на мембраната си и не стимулират вродения имунен отговор, некротичните клетки стават пермеабилизирани и секретират ендогенни адюванти, които могат да предизвикат силен вроден имунен отговор. [ 60 ]
Тъй като нивото на апоптоза/некроза корелира с имуногенността на ротавируса, се предполага, че имуностимулиращият ефект на апоптотичните/некротичните клетки най-вероятно допринася за генерирането на защитен имунен отговор. Следователно, регулирането на експресията на ротавирусния гама-резистентност (RV G) е много вероятно важен фактор в патогенезата на бяса, тъй като осигурява средство за оцеляване и разпространение на патогенни варианти на RV в нервната система, без да причинява явно невронално увреждане и да предизвиква защитен имунен отговор, който би предотвратил инфекцията.
Експресията на RV G може да бъде регулирана на ниво синтез на РНК, посттранслационно ниво или и двете. Нивата на RV G, експресирани от различни RV химерни варианти, са показали, че се отразяват от скоростта на синтез на вирусна РНК, което предполага, че диференциалната регулация на експресията на RV G от тези варианти е резултат от вариации в скоростта на транскрипция на вирусна мРНК. Както при скоростите на транскрипция на вирусна РНК, количеството RV G, експресирано от тези варианти, е обратнопропорционално корелирано с вирусната патогенност. От друга страна, инфекцията на първични невронни култури с по-малко патогенния RV вариант CVS-B2c води до четири пъти по-високи нива на G протеин, отколкото инфекцията с високопатогенния вариант CVS-N2c, въпреки синтеза на сравними нива на G мРНК и при двете инфекции. Експериментите с импулсно преследване показват, че по-високите нива на G протеин в невроните, инфектирани с CVS-B2c, са до голяма степен резултат от по-ниска скорост на разграждане на G протеина CVS-B2c в сравнение с G протеина CVS-N2c. Въпреки това, механизмът, който води до по-бързото протеолитично разграждане на CVS-N2c G протеина, все още не е изяснен.
Симптоми бяс
Инкубационният период при бяс е средно 30-90 дни. В случай на масивна инфекция през големи рани по главата и лицето, той може да бъде съкратен до 12 дни. В редки случаи инкубационният период може да продължи 1 година или повече.
Наблюдава се строго последователна промяна на три периода на заболяването: продромален, възбуден, паралитичен.
Продромалният период започва с появата на болезнена или дърпаща болка на мястото на ухапване, както и болка по хода на нервите. В областта на белега може да се появи усещане за парене, сърбеж, понякога зачервяване и подуване. Пациентът изпитва общо неразположение, главоболие, гадене. Отбелязват се повръщане, повишаване на телесната температура до 37,5-38°C и симптоми на прогресиращо психично разстройство: повишена рефлекторна възбудимост, необяснимо чувство на тревожност, страх, меланхолия. Често пациентът е депресиран, потиснат, затворен, отказва да се храни, спи лошо, оплаква се от мрачни мисли, плашещи сънища. Продромалният период продължава 2-3 дни, понякога се удължава до 7 дни. В края на този период могат да се появят пристъпи на тревожност с краткотрайни затруднения в дишането, усещане за стягане в гърдите, придружени от тахикардия и учестено дишане.
Периодът на възбуда се характеризира с появата на хидрофобия: при опит за пиене, а след това и при вида на вода или напомняне за нея, пациентът изпитва конвулсивен спазъм на фаринкса и ларинкса, по време на който изхвърля халбата с вода с писък, хвърля напред треперещи ръце, отмята глава и тяло назад. Вратът е изпънат, болезнена гримаса изкривява лицето, което става синкаво поради спазъм на дихателните мускули. Очите изпъкват, изразяват страх, молят за помощ, зениците са разширени, вдишването е затруднено. В разгара на атаката е възможен сърдечен и дихателен арест. Атаката продължава няколко секунди, след което състоянието на пациента сякаш се подобрява. Впоследствие могат да се появят пристъпи на спазми на мускулите на ларинкса и фаринкса дори от движението на въздуха (аерофобия), ярка светлина (фотофобия) или силна дума (акустикофобия). Пристъпите са съпроводени с психомоторна възбуда, по време на която пациентът се държи като „луд“. Съзнанието е замъглено по време на пристъпа, но се прояснява в интерикталния период. В периода на възбуда, поради повишения тонус на симпатиковата нервна система, пациентите изпитват рязко повишаване на слюноотделянето (сиалорея) с невъзможност за преглъщане на слюнка поради спазъм на фарингеалните мускули. Пациентът пръска слюнка. Някои пациенти могат да развият признаци на менингизъм и дори опистотонус, а гърчовете са чести. В този случай цереброспиналната течност може да не се промени, но при някои пациенти концентрацията на протеини може да се увеличи, а броят на клетките може да се увеличи поради лимфоцитите.
Без адекватно лечение, признаците на дехидратация се засилват, чертите на лицето стават по-остри, а телесното тегло намалява. Телесната температура се повишава до високи стойности. Възможни са конвулсии. Продължителността на стадия на възбуждане е около 2-3 дни, рядко 4-5 дни. Фатален изход обикновено настъпва по време на един от пристъпите. Рядко пациентът оцелява до третия стадий на заболяването.
По време на периода на парализа пациентът се успокоява. Пристъпите на хидрофобия спират, пациентът може да пие и преглъща храна, съзнанието е ясно. Въпреки привидното благополучие обаче, летаргията, апатията, депресията се засилват, скоро се появяват парализа на крайниците, тазови нарушения, парализа на черепномозъчните нерви. Телесната температура се повишава до 42-43°C, артериалното налягане спада и до края на първия ден настъпва смърт от парализа на сърдечно-съдовия и дихателния център.
В периферната кръв се наблюдават неутрофилна левкоцитоза, повишен хемоглобин, еритроцити и хематокрит.
Какво те притеснява?
Форми
Клинично се разграничават типични и атипични форми. Атипичните форми включват всички случаи без възбуда и хидрофобия. Атипичните форми включват булбарна, церебеларна, менингоенцефалитна и др.
Диагностика бяс
Откриването на антиген на бяс, антитела, вирусна РНК или изолиране на вируса позволява диагностициране на бяс. Тъй като всеки отделен тест може да е отрицателен при пациент с бяс, понякога са необходими серийни серумни проби за откриване на антитела срещу бяс, проби от слюнка за вирусна култура и кожна биопсия за директно имунофлуоресцентно изследване за вирусен антиген, особено когато има силно съмнение за бяс.
Един от най-бързите методи за диагностициране на бяс преди смъртта при хора е извършването на директен имунофлуоресцентен тест върху кожна биопсия от тила за откриване на антиген на бяс. Директният имунофлуоресцентен тест е най-чувствителният и специфичен метод за откриване на антиген на бяс в кожа и други пресни тъкани (напр. мозъчна биопсия), въпреки че резултатите понякога могат да бъдат отрицателни в началото на заболяването. Ако прясна тъкан не е налична, ензимното разграждане на фиксирани тъкани може да увеличи реактивността на имунофлуоресцентния тест; чувствителността обаче може да бъде неприемливо ниска.
Диагнозата може да се постави и ако вирусът се изолира от слюнка след инокулация на невробластомни клетки или лабораторни гризачи; това обикновено е най-ефективно през първите 2-3 седмици от заболяването. Откриването на антитела, неутрализиращи вируса на бяс, обикновено извършвано чрез бърз флуоресцентен фокусен инхибиторен тест (RFFIT), в серума на неваксинирани индивиди също е диагностично. Наличието на антитела в цереброспиналната течност потвърждава диагнозата, но те могат да се появят 2-3 дни по-късно от серумните антитела и следователно може да са по-малко полезни в ранните стадии на заболяването. Докато серологичният отговор след ваксинацията обикновено е неразличим от серологичния отговор, предизвикан от заболяването, ваксинацията обикновено не произвежда антитела към цереброспиналната течност.
Само седем случая на „възстановяване“ от бяс през последните 25 години са добре документирани. Въпреки че вирусът на бяс не е изолиран от нито един от пациентите, високите титри на антитела, неутрализиращи бяс, в серумни проби и наличието на неутрализиращи антитела в цереброспиналната течност категорично подкрепят диагнозата.
Какво трябва да проучим?
Какви тестове са необходими?
Диференциална диагноза
Диагнозата бяс при хората обикновено се поставя въз основа на епидемиологични и клинични данни и се потвърждава лабораторно. Диагнозата е лесна за поставяне, ако има анамнеза за ухапвания от животни и е налице пълният спектър от симптоми и признаци. В противен случай е необходима внимателна, но бърза оценка на епидемиологичните и клиничните характеристики на по-рядко типичните случаи, преди да се извършат специфични лабораторни изследвания. Всеки пациент с неврологични признаци или симптоми или необясним енцефалит трябва да бъде разпитан за възможността за контакт с животни в ендемични за бяс райони в или извън страната на пребиваване. Неподозирането за бяс при няколко скорошни смъртни случая на хора в Съединените щати може да се дължи на липса на внимателна анамнеза за контакт.
В началото на заболяването, бясът може да имитира много инфекциозни и неинфекциозни заболявания. Много други енцефалити, като тези, причинени от херпесни вируси и арбовируси, наподобяват бяс. Други инфекциозни заболявания също могат да имитират бяс, като тетанус, церебрална малария, рикетсиоза и коремен тиф. Паралитичните инфекциозни заболявания, които могат да бъдат объркани с бяс, включват полиомиелит, ботулизъм и херпесен маймунски енцефалит B.
Неинфекциозните заболявания, които могат да бъдат объркани с бяс, включват редица неврологични синдроми, особено остра възпалителна полиневропатия (синдром на Гилен-Баре), както и алергичен постваксинален енцефаломиелит, вторичен на ваксинация срещу бяс на нервната тъкан, отравяне или наркотична интоксикация, абстиненция от алкохол, остра порфирия и бясна истерия. Синдромът на Гилен-Баре може да бъде объркан с паралитичен бяс и обратно.
Към кого да се свържете?
Лечение бяс
Лечението на бяс не е разработено. Прилагането на големи дози специфичен антирабичен имуноглобулин и левкоцитен интерферон е неефективно. За облекчаване на страданието на пациента се прилага симптоматично лечение. За тази цел пациентът се настанява в отделно отделение или бокс, създава се защитен режим, който ограничава влиянието на външната среда (намален шум, ярка светлина, въздушен поток). За намаляване на възбудимостта на централната нервна система се предписват сънотворни, антиконвулсанти и болкоуспокояващи. Водният баланс се нормализира.
В паралитичния стадий се предписват лекарства, които стимулират активността на сърдечно-съдовата и дихателната система. Препоръчва се прилагането на хипербарна оксигенация, церебрална хипотермия, контролирано механично дишане с пълна кураризация на пациента. Всички методи на лечение обаче са практически неефективни. В най-добрия случай е възможно да се удължи животът на пациента с няколко месеца. Неблагоприятният изход е предопределен от тежестта на увреждането на мозъчния ствол с разрушаване на жизненоважни центрове.
Предотвратяване
Разработването на първата ваксина срещу бяс от Пастьор през 1885 г. въвежда ерата на много по-ефективен контрол на бяса. Днес, въпреки почти 100% смъртност при хората от бяс, заболяването е напълно предотвратимо чрез ваксинация преди и/или след експозиция. Докато Пастьор и неговите колеги инициират ваксинацията на частни кучета в Париж, първата масова ваксинация на кучета е проведена в началото на 20-те години на миналия век в Япония, отбелязвайки първата голяма национална програма за контрол на бяса. Оралната ваксинация на диви животни, разработена за първи път през 70-те години на миналия век, оттогава многократно е доказано, че ефективно контролира заболяването при основни сухоземни гостоприемници като лисици, миещи мечки и скунксове.[ 68 ] Продължителната ваксинация срещу бяс на популациите от резервоарни животни при 70% или по-високи нива на покритие в крайна сметка ще елиминира RABV от резервоарните видове и ще предотврати разпространението на вируса към случайни гостоприемници.[ 69 ]
Филогенетичните данни показват, че лисавирусите са заразили прилепите много преди да заразят сухоземните бозайници, а повечето лисавируси, включително RABV, все още циркулират в различни видове прилепи по целия свят.[ 70 ] Въпреки това, ефективни методи за предотвратяване на предаването на RABV сред прилепите остават неуловими, което изключва възможността за пълно унищожаване на бяса в този момент. Въпреки това, дори след излагане на RABV чрез ухапване от заразен с бяс бозайник, безопасната и ефективна постекспозиционна профилактика (PEP, включително почистване на рани, имуноглобулин срещу бяс и ваксинация срещу бяс) може да предпази хората от инфекция с бяс, ако лечението се приложи своевременно и съгласно препоръките на Световната здравна организация (СЗО).
Тези два метода за предотвратяване на човешки смъртни случаи – единият, основан на ваксиниране на изложени на вируса хора, а другият, основан на ваксиниране на достатъчно кучета, за да се прекъсне цикълът на предаване при източника – са градивните елементи на подхода „едно здраве“ за превенция и контрол на бяса при кучетата. Тези два различни начина за предотвратяване на човешки смъртни случаи бяха разгледани като отделни алтернативи: Стратегия А, основана на предоставяне на PEP на хора, и Стратегия Б, основана на ваксиниране на кучета; или като компоненти на комбинирана Стратегия А + Б при анализ на вероятните разходи за алтернативните стратегии.[ 71 ]
Страни като Тайланд са постигнали огромен успех в предотвратяването на човешки смъртни случаи чрез използването на PEP, но също така са установили нарастващо търсене и свързани с него разходи, свързани само с употребата на PEP. [ 72 ] Например, в сравнение със ситуацията през 1991 г., четири пъти повече хора (повече от 400 000) са се нуждаели от PEP през 2003 г. Последните данни показват, че Китайската народна република, която ваксинира 15 милиона души годишно след потенциално излагане на бяс, харчи около 650 милиона щатски долара годишно само за PEP. [ 73 ]
Много по-устойчив подход е да се предотврати разпространението на инфекцията в източника, в животинската популация, като същевременно се увеличи достъпът до PEP за изложени на инфекцията човешки пациенти, когато е необходимо. Където има политическа воля и адекватно финансиране за контрол на бяса при кучетата, смъртните случаи могат и са били елиминирани. Широко разпространеното използване на ваксинация на кучета доведе до елиминирането на бяса при кучетата в няколко страни, включително Малайзия през 1954 г., [ 74 ] Япония през 1956 г., Тайван през 1961 г., Сингапур и по-специално в цяла Западна Европа (прегледано в Rupprecht et al., King et al., и Gongal and Wright). [ 75 ]