
Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Лисенцефалия на мозъка
Медицински експерт на статията

Последно прегледани: 12.07.2025
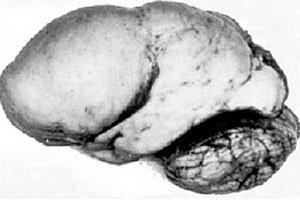
Сред органичните мозъчни патологии се откроява вродена аномалия в развитието на мозъка като лизенцефалията, чиято същност се крие в почти гладката повърхност на кората на нейните полукълба - с недостатъчен брой извивки и бразди. [ 1 ]
При пълна липса на извивки се определя агирия, а наличието на няколко широки плоски извивки се нарича пахигирия. Тези дефекти, както и някои други редукционни деформации на мозъка, имат код Q04.3 в МКБ-10.
Епидемиология
Според статистиката за редките заболявания, има 1-1,2 случая на лизенцефалия на 100 хиляди новородени. [ 2 ], [ 3 ]
Според някои данни, до 25-30% от случаите на класическа лизенцефалия се наблюдават при деца със синдром на Милър-Дикер; точкови мутации и делеции на гените LIS1 и DCX се откриват при почти 85% от пациентите. [ 4 ]
Генетични изследвания на 17 гена, свързани с лизенцефалия, показват, че мутацията или делецията на LIS1 е причина за 40% от пациентите, а 23% са свързани с DCX мутация, следвани от TUBA1A (5%) и DYNC1H1 (3%).[ 5 ]
Причини Лисенцефалия
Всички известни причини за образуването на мозъчната кора (cortex cerebri) почти или напълно без извивки и бразди, които увеличават „работната площ“ на човешкия мозък и осигуряват „продуктивността“ на централната нервна система, са свързани с нарушения в нейното перинаталното развитие. Тоест, лизенцефалията се развива при плода. [ 6 ]
Невъзможността за формиране на слоеве от мозъчната кора на плода при лизенцефалия е резултат от анормална миграция на невроните, които я образуват, или преждевременно прекратяване на този процес.
Този процес, който е от съществено значение за цереброкортикалната хистогенеза, протича на няколко етапа от 7-та до 18-та седмица от бременността. И предвид повишената му чувствителност към генетични мутации, както и различни негативни физични, химични и биологични влияния, всяко отклонение от нормата може да доведе до неправилна локализация на невроните с евентуално образуване на удебелен слой сиво вещество на кората без характерна структура. [ 7 ]
В някои случаи лизенцефалията при деца е свързана със синдроми на Милър-Дикер, Уокър-Варбург или Норман-Робъртс.
Прочетете също – Дефекти в развитието на мозъка
Рискови фактори
В допълнение към мутациите в някои гени, рисковите фактори за раждането на дете с такъв тежък дефект включват кислородно гладуване (хипоксия) на плода; недостатъчно кръвоснабдяване на мозъка (хипоперфузия); остър мозъчносъдов инцидент под формата на перинатален инсулт; плацентарни патологии; вирусни инфекции на бременната жена (включително TORCH); [ 8 ] проблеми с общия метаболизъм и функцията на щитовидната жлеза; тютюнопушене, алкохол, психотропни и наркотични вещества; употреба на редица лекарства; повишени нива на радиация. [ 9 ]
Патогенеза
Не всички случаи на лизенцефалия имат патогенеза, причинена от хромозомни аномалии и генни мутации. Но са известни някои гени, които кодират протеини, играещи важна роля в правилното движение на невробластите и невроните по радиалните глия клетки - за да образуват мозъчната кора. А мутациите на тези гени водят до тази патология. [ 10 ]
По-специално, това са спорадични мутации (без наследственост) на гена LIS1 на хромозома 17, който регулира цитоплазмения двигателен протеин на микротубулите динеин, както и гена DCX на Х хромозомата, който кодира протеина двойнокортин (лисенцефалин-X). [ 11 ]. В първия случай специалистите определят класическа лисенцефалия (тип I), във втория – Х-свързана. [ 12 ]
Когато генът FLN1, който кодира фосфопротеина филамин 1, бъде премахнат, процесът на насочена невронна миграция може изобщо да не започне, което води до пълна липса на извивки (агирия). [ 13 ]
Идентифицирани са мутации в гена CDK5, който кодира киназен ензим – катализатор за вътреклетъчния метаболизъм, регулиращ клетъчния цикъл в невроните на ЦНС и осигуряващ нормалната им миграция по време на пренаталното формиране на мозъчните структури.
Анормалните промени в гена RELN на хромозома 7, които причиняват кортикални гирални дефекти при синдрома на Норман-Робъртс, водят до дефицит на извънклетъчния гликопротеин реелин, който е необходим за регулиране на миграцията и позиционирането на невралните стволови клетки по време на развитието на мозъчната кора.[ 14 ], [ 15 ], [ 16 ]
Генът ARX кодира неаристаленовия хомеобокс протеин, транскрипционен фактор, който играе важна роля в предния мозък и други тъкани.[ 17 ] Децата с ARX мутацията имат други симптоми, като липсващи части от мозъка (агенезия на corpus callosum), анормални гениталии и тежка епилепсия.[ 18 ],[ 19 ]
Няколко гена са свързани с лизенцефалия. Тези гени включват VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A и CDK5.[ 20 ]
Цитомегаловирусът (CMV) е свързан с развитието на лизенцефалия поради намалено кръвоснабдяване на мозъка на плода. Тежестта на CMV инфекцията зависи от гестационната възраст. Ранната инфекция е по-вероятно да причини лизенцефалия, тъй като невронната миграция се случва в ранен етап на бременността.[ 21 ]
Освен това, механизмът на възникване на тази аномалия включва непълно или по-късно спиране на миграцията на неврони от перивентрикуларната генеративна зона към мозъчната кора. И в такива случаи се развива или непълна лизенцефалия, или пахигирия, при която се образуват няколко широки жлеба и извивки (но повечето от тях липсват).
Симптоми Лисенцефалия
Първите признаци на тази патология (при липса на споменатите по-рано синдроми) може да не се появят веднага след раждането, а след месец и половина до два. И най-често се наблюдават следните клинични симптоми на лизенцефалия:
- мускулна хипотония, често комбинирана със спастична парализа;
- конвулсии и генерализирани тонично-клонични припадъци (под формата на опистотонус);
- тежка умствена изостаналост и забавяне на растежа;
- нарушаване на неврологичните и двигателните функции.
Проблемите с преглъщането причиняват трудности при храненето на бебето. [ 22 ]
Високата степен на невромоторно увреждане често се проявява като тетраплегия – парализа на всички крайници. Възможна е деформация на ръцете, пръстите на ръцете или краката.
При синдрома на Норман-Робъртс с лизенцефалия тип I се наблюдават краниофациални аномалии: тежка микроцефалия, нисък наклон на челото и изпъкнал широк мост на носа, широко разположени очи (хипертерлоризъм), недоразвитие на челюстите (микрогнатия). [ 23 ]
Синдромът на Милър-Дикер може да се характеризира и с необичайно малък размер на главата с широко, високо чело и къс нос, вдлъбнатини в слепоочията (битемпорални вдлъбнатини) и ниско разположени, деформирани уши.
Тежкият синдром на лизенцефалия се характеризира с микроцефалия, намаляване на размера на очните ябълки (микрофталмия), комбинирано с ретинална дисплазия, обструктивна хидроцефалия и липсващ или хипопластичен corpus callosum.
Усложнения и последствия
Сред усложненията на тази аномалия, специалистите посочват нарушение на преглъщащата функция (дисфагия) и гастроезофагеален рефлукс; рефрактерна (неконтролирана) епилепсия; чести инфекции на горните дихателни пътища; пневмония (включително хронична аспирация).
Кърмачетата с лизенцефалия могат да имат вродени органични сърдечни проблеми под формата на дефект на междупредсърдната преграда или сложен сърдечен дефект с цианоза (тетралогия на Фало). [ 24 ]
Последиците от постнаталното забавяне на растежа в повечето случаи водят до смърт в рамките на 24 месеца след раждането.
Диагностика Лисенцефалия
Диагнозата започва с физически преглед на детето, проучване на медицинската история на родителите и историята на бременността и раждането.
По време на бременността може да се наложи безклетъчно изследване на фетална ДНК, амниоцентеза или вземане на хорионни въси. [ 25 ] За повече информация вижте – Пренатална диагностика на вродени заболявания
Инструменталната диагностика се използва за визуализиране на мозъчните структури и оценка на техните функции:
- компютърна томография на мозъка;
- магнитно-резонансна томография (ЯМР) на мозъка;
- електроенцефалограма (ЕЕГ). [ 26 ]
По време на бременност, лизенцефалията при ултразвуково изследване на плода след 20-21 седмици може да се подозира при липса на парието-тилни и калкарни жлебове и аномалия на Силвиевата фисура на мозъка.
Диференциална диагноза
Провежда се диференциална диагностика с други синдроми на вродени мозъчни дефекти.
Съществуват повече от 20 вида лизенцефалия, повечето от които попадат в 2 основни категории: класическа лизенцефалия (тип 1) и лизенцефалия тип „каменна кухина“ (тип 2). Всяка категория има сходни клинични прояви, но различни генетични мутации.[ 27 ]
Изследването на мозъка при лизенцефалия тип I показва мозъчна кора с четири слоя вместо шест, както при нормалните пациенти, докато при лизенцефалия тип 2 мозъчната кора е дезорганизирана и изглежда бучковата или нодуларна поради пълното ѝ изместване от клъстери от кортикални неврони, разделени от глиомезенхимна тъкан. Пациентите също така са имали мускулни и очни аномалии.
- Класическа лизенцефалия (тип 1):
- LIS1: Изолирана лизенцефалия и синдром на Милър-Дикер (лизенцефалия, свързана с лицев дисморфизъм). [ 28 ]
- LISX1: мутация в гена DCX. В сравнение с лизенцефалията, причинена от мутации в LIS1, DCX показва шестслойна кора вместо четири.
- Изолирана лизенцефалия без други известни генетични дефекти
- Лисенцефалия тип „калдъръм“ (тип 2):
- Синдром на Уокър-Варбург
- Синдром на Фукуяма
- Болест на мускулите, очите и мозъка
- Други видове не могат да бъдат поставени в една от двете горни групи:
- LIS2: Синдром на Норман-Робъртс, подобен на лизенцефалия тип I или синдром на Милър-Дикер, но без делеция на хромозома 17.
- LIS3
- LISX2
Микролисенцефалия: Това е комбинация от липса на нормално кортикално сгъване и необичайно малка глава. Децата с правилна лисенцефалия имат нормален размер на главите при раждане. Децата с малък размер на главата при раждане обикновено се диагностицират с микролисенцефалия.
Важно е също да се прави разлика между лизенцефалия и полимикрогирия, които са различни дефекти в развитието на мозъка.
Към кого да се свържете?
Лечение Лисенцефалия
Лизенцефалията е нелечим органичен дефект, така че е възможно само поддържащо и симптоматично лечение. [ 29 ]
На първо място, това е употребата на антиконвулсанти и антиепилептични лекарства, както и поставянето на гастростомична тръба в стомаха (ако детето не е в състояние да преглъща самостоятелно). Масажът е полезен.
В случаи на тежка хидроцефалия се отстранява цереброспиналната течност.
Предотвратяване
Експертите препоръчват бъдещите родители да потърсят генетична консултация, а бременните жени своевременно да се регистрират при акушер-гинеколози и да се подложат на всички планирани прегледи.
Прогноза
При деца с лизенцефалия прогнозата зависи от степента ѝ, но най-често психическото развитие на детето не надвишава нивото от четири до пет месеца. И всички деца с тази диагноза страдат от тежки психомоторни нарушения и труднолечима епилепсия. [ 30 ]
Според NINDS (Американският национален институт по неврологични разстройства и инсулт), максималната продължителност на живота за хора с лизенцефалия е около 10 години.