
Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Синдром на Ашер
Медицински експерт на статията

Последно прегледани: 04.07.2025
Синдромът на Ушър е наследствено заболяване, което се проявява като пълна глухота от раждането, както и като прогресивна слепота с възрастта. Загубата на зрение е свързана с пигментозен ретинит, процес на пигментна дегенерация на ретината. Много хора със синдром на Ушър имат и тежки проблеми с равновесието.
Епидемиология
Благодарение на изследването беше възможно да се установи, че синдромът на Ушер засяга около 8% от изследваните глухонеми деца (тестовете са проведени в специални институции за глухонеми хора). Пигментен ретинит е наблюдаван при 6-10% от пациентите, страдащи от вродена глухота, която от своя страна се наблюдава при около 30% от хората с пигментно заболяване на ретината.
Смята се, че това заболяване се проявява при приблизително 3-10 души от 100 хиляди в световен мащаб. Може да се наблюдава както при жени, така и при мъже еднакво. Около 5-6% от световното население страда от този синдром. Около 10% от всички случаи на дълбока глухота в детска възраст се дължат на синдром на Usher I, както и на II тип.
В Съединените щати, типове 1 и 2 са най-често срещаните. Заедно те представляват приблизително 90 до 95 процента от всички случаи на синдром на Ушер при деца.
Причини Синдром на Ашер
Синдромът на Ушер тип I, II и III има автозомно-рецесивен произход, докато тип IV се счита за Х-хромозомно разстройство. Причините за слепота и глухота, които се срещат при този синдром, все още не са достатъчно проучени. Предполага се, че хората с това заболяване са свръхчувствителни към компоненти, които могат да увредят структурата на ДНК. Освен това, това заболяване може да е свързано с нарушения на имунната система, но в този случай няма точна картина на този процес.
През 1989 г. хромозомни аномалии са идентифицирани за първи път при пациенти с заболяване тип II, което в бъдеще може да доведе до начин за изолиране на гените, причиняващи синдрома. Възможно е също така да се идентифицират тези гени при носителите и да се разработят специални пренатални генетични тестове.
 [ 8 ]
[ 8 ]
Рискови фактори
Синдромът се наследява, когато и двамата родители са засегнати, т.е. унаследява се по рецесивен тип. Дете може да наследи заболяването и ако родителите му са носители на гена. Ако и двамата бъдещи родители имат този ген, тогава вероятността да се роди бебе с този синдром е 1 към 4. Човек, който има само един ген за синдрома, се счита за носител, но няма симптоми на заболяването. В днешно време все още не е възможно да се определи дали човек има гена за това заболяване.
Ако дете се роди от родители, единият от които няма такъв ген, тогава вероятността то да наследи синдрома е много ниска, но определено ще бъде носител.
Симптоми Синдром на Ашер
Симптомите на синдрома на Ушър включват загуба на слуха и анормално натрупване на пигментирани клетки в очните структури. След това пациентът развива дегенерация на ретината, което причинява влошаване на зрението и евентуална загуба на зрение в най-тежките случаи.
Сензорневралната загуба на слуха може да бъде лека или пълна и обикновено не прогресира от раждането. Пигментното заболяване на ретината обаче може да започне да се развива в детството или по-късно. Резултатите от тестовете показват, че централната зрителна острота може да се поддържа в продължение на много години, дори когато периферното зрение се влоши (състояние, наречено „тунелно зрение“).
Това са основните прояви на заболяването, които понякога могат да бъдат допълнени от други разстройства, като психоза и други психични разстройства, проблеми с вътрешното ухо и/или катаракта.
Форми
По време на изследването са идентифицирани 3 вида на това заболяване, както и 4-та форма, която е доста рядка.
Тип I на заболяването се характеризира с вродена пълна глухота, както и с нарушение на равновесието. Често такива деца започват да ходят едва на 1,5 години. Влошаването на зрението обикновено започва на 10-годишна възраст, а окончателното развитие на състоянието на нощна слепота започва на 20-годишна възраст. Децата с този вид заболяване могат да развият прогресивно влошаване на периферното зрение.
При заболяване тип II се наблюдава умерена или вродена глухота. В този случай влошаването на частичната глухота често не се наблюдава. Пигментният ретинит започва да се развива около края на юношеството или след 20 години. Развитието на нощната слепота обикновено започва на 29-31 години. Нарушението на зрителната острота при патология тип II обикновено прогресира малко по-бавно, отколкото при тип I.
Тип III на заболяването се характеризира с прогресивна загуба на слуха, обикновено започваща по време на пубертета, както и с постепенното развитие през същия период (малко по-късно от загубата на слуха) на пигментозен ретинит, който може да се превърне във фактор за развитието на прогресивна слепота.
Проявите на патология тип IV се срещат главно при мъжете. В този случай се наблюдават и прогресивни нарушения и загуба на слуха и зрението. Тази форма е много рядка и обикновено има Х-хромозомна природа.
Диагностика Синдром на Ашер
Диагнозата на синдрома на Ушер се поставя въз основа на наблюдаваната при пациента комбинация от внезапна глухота и прогресивна загуба на зрение.
Тестове
Може да бъде назначен специален генетичен тест за откриване на мутацията.
Открити са единадесет генетични локуса, които могат да причинят развитието на синдрома на Ушер, и са идентифицирани девет гена, които определено са причината за разстройството:
- Тип 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Тип 2: ush2a, VLGR1, WHRN.
- Синдром на Ушер тип 3: USH3A.
Учени от NIDCD, заедно с колеги от университети в Ню Йорк и Израел, са идентифицирали мутация, наречена R245X, в гена Pcdh15, която е причина за голям процент от случаите на синдром на Usher тип 1 сред еврейското население.
За да научите повече за лаборатории, които извършват клинични изпитвания, посетете https://www.genetests.org и потърсете в директорията на лабораториите „синдром на Ушер“.
За да научите за съществуващите клинични изпитвания, които включват генетично тестване за синдром на Usher, посетете https://www.clinicaltrials.gov и потърсете „Usher syndrome“ или „Usher syndrome genetic testing“.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Инструментална диагностика
Има няколко метода за инструментална диагностика:
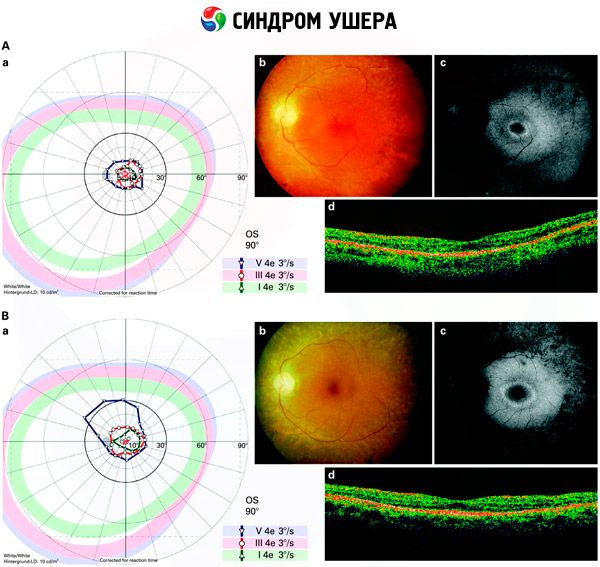
- Изследване на фундуса за откриване на наличието на пигментни петна по ретината, както и стесняване на съдовете на ретината;
- Електроретинограма, която позволява да се открият начални дегенеративни отклонения в ретината на окото. Тя показва изчезване на електрорентгенографските пътища;
- Електронистагмографията (ЕНГ) измерва неволеви движения на очите, които биха могли да показват наличието на дисбаланс.
- Аудиометрия, която се използва за определяне на наличието на глухота и нейната тежест.
Диференциална диагноза
Синдромът на Ушер трябва да се диференцира от някои подобни разстройства.
Синдром на Халгрен, който се характеризира с вродена загуба на слуха и прогресивна загуба на зрението (развиват се също катаракта и нистагъм). Допълнителни симптоми включват атаксия, психомоторни нарушения, психоза и умствена изостаналост.
Синдром на Алстром, който е наследствено заболяване, при което ретината дегенерира, което води до загуба на централно зрение. Този синдром е свързан с детското затлъстяване. В същото време, захарният диабет и загубата на слуха започват да се развиват след 10 години.
Рубеолата при бременна жена през първия триместър може да причини различни аномалии в развитието на детето. Сред последствията от такава аномалия са загуба на слуха, както и (или) проблеми със зрението, а освен това и различни дефекти в развитието.
Към кого да се свържете?
Лечение Синдром на Ашер
В момента няма лечение за синдрома на Ушър. Следователно, терапията в този случай се състои главно в забавяне на процеса на загуба на зрение, както и в компенсиране на загубата на слуха. Възможните методи на лечение включват:
- Прием на витамин А (някои офталмолози смятат, че високите дози витамин А палмитат могат да забавят, но не и да спрат, прогресията на пигментозния ретинит);
- Имплантиране на специални електронни устройства в ушите на пациента (слухови апарати, кохлеарни импланти).
Офталмолозите препоръчват на повечето възрастни с често срещани форми на пигментозен ретинит да приемат 15 000 IU (международни единици) витамин А палмитат дневно под наблюдение. Тъй като хора със синдром на Usher тип 1 не са били включени в проучването, високи дози витамин А не се препоръчват за тази група пациенти. Хората, които обмислят прием на витамин А, трябва да обсъдят тази възможност за лечение с лекаря си. Други препоръки за тази възможност за лечение включват:
- Променете диетата си, като включите храни, богати на витамин А.
- Жените, които планират бременност, трябва да спрат приема на високи дози витамин А три месеца преди планираното зачеване, поради повишен риск от вродени дефекти.
- Бременните жени трябва да спрат приема на високи дози витамин А поради повишен риск от вродени дефекти.
Важно е също така такова дете да се адаптира към социалния живот. Това изисква помощта на специални педагоги и психолози. В случай че пациентът е започнал да изпитва прогресивна загуба на зрение, той трябва да бъде научен да използва жестомимичен език.
Прогноза
Синдромът на Ушер има неблагоприятна прогноза. Зрителното поле и неговата острота започват да се влошават в периода от 20-30 години при повечето пациенти с това заболяване от всякакъв вид. В някои случаи настъпва пълна двустранна загуба на зрение. Загубата на слуха, която винаги е съпроводена с глухота, много бързо се развива до пълна двустранна загуба на слуха.