
Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Болест на Шарко-Мари-Тоот.
Медицински експерт на статията

Последно прегледани: 12.07.2025
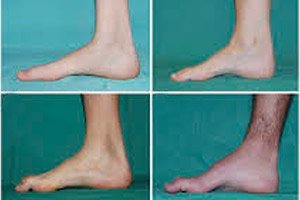
Перонеалната мускулна атрофия, синдром или болест на Шарко-Мари-Тут, е група от хронични наследствени заболявания с увреждане на периферните нерви.
Според МКБ-10, в раздела за заболявания на нервната система, кодът за това заболяване е G60.0 (наследствена двигателна и сензорна невропатия). То е включено и в списъка на орфанните заболявания.
Епидемиология
Според клиничната статистика, разпространението на всички видове болест на Шарко-Мари-Тут на 100 хиляди от населението е 19 случая (според други източници, един случай на 2,5-10 хиляди от населението).
CMT тип 1 представлява около две трети от случаите (един случай на 5000 до 7000 души от населението), като близо 70% от тях са свързани с дублиране на гена PMP22. Повече от 1,2 милиона души по света страдат от този вид заболяване.
Честотата на CMT тип 4 се оценява на 1-5 случая на 10 000 деца. [ 1 ]
Причини Болест на Шарко-Мари-Тоот
Според класификацията на полиневропатичните синдроми, перонеалната (фибуларна) мускулна атрофия, невронната амиотрофия на Шарко-Мари-Тут или болестта на Шарко-Мари-Тут (съкратено CMT) се отнася до генетично обусловени моторно-сензорни полиневропатии. [ 2 ]
Тоест, причините за възникването му са генетични мутации. И в зависимост от естеството на генетичните отклонения се разграничават основните видове или видове този синдром: демиелинизиращ и аксонален. Първата група включва болестта на Шарко-Мари-Тут тип 1 (CMT1), която възниква поради дублиране на гена PMP22 на хромозома 17, който кодира трансмембранния периферен миелинов протеин 22. В резултат на това настъпва сегментна демиелинизация на аксоновата обвивка (израстъците на нервните клетки) и намаляване на скоростта на провеждане на нервния сигнал. Освен това, мутации могат да бъдат и в някои други гени.
Аксоналната форма е болест на Шарко-Мари-Тут тип 2 (CMT2), която засяга самите аксони и е свързана с патологични промени в гена MFN2 в локус 1p36.22, кодиращ мембранния протеин митофузин-2, който е необходим за сливането на митохондриите и образуването на функционални митохондриални мрежи вътре в периферните нервни клетки. Съществуват повече от дузина подтипове на CMT2 (с мутации в специфични гени).
Трябва да се отбележи, че понастоящем са идентифицирани повече от сто гена, чието увреждане, предавано по наследство, причинява различни подтипове на болестта на Шарко-Мари-Тут. Например, мутациите в гена RAB7 водят до CMT тип 2B; промяната в гена SH3TC2 (кодиращ един от мембранните протеини на клетките на Шван) причинява CMT тип 4C, която се проявява в детска възраст и се характеризира с демиелинизация на двигателните и сензорните неврони (съществуват дузина и половина форми на тип 4 на това заболяване).
Рядката CMT тип 3 (наречена синдром на Дежерин-Сотас) започва да се развива в ранна детска възраст и се причинява от мутации в гените PMP22, MPZ, EGR2 и други.
Когато CMT тип 5 се появи на възраст 5-12 години, се наблюдава не само двигателна невропатия (под формата на спастична парапареза на долните крайници), но и увреждане на зрителния и слуховия нерв.
Мускулната слабост и оптичната атрофия (със загуба на зрение), както и проблемите с равновесието са типични за CMT тип 6. А при болестта на Charcot-Marie-Tooth тип 7 има не само моторно-сензорна невропатия, но и заболяване на ретината под формата на пигментозен ретинит.
По-често срещана сред мъжете, Х-свързаната CMT или болестта на Шарко-Мари-Тут с тетрапареза на крайниците (слабост на движението както на ръцете, така и на краката) е демиелинизиращ тип и се смята, че е резултат от мутация в гена GJB1 на дългото рамо на Х хромозомата, който кодира конексин 32, трансмембранен протеин на клетките на Шван и олигодендроцитите, който регулира предаването на нервни сигнали. [ 3 ]
Рискови фактори
Основният рисков фактор за CMT е фамилната анамнеза за заболяването, т.е. при близки роднини.
Според генетиците, ако и двамата родители са носители на автозомно-рецесивния ген за болестта на Шарко-Мари-Тут, рискът от раждане на дете, което ще развие това заболяване, е 25%. А рискът детето да бъде носител на този ген (но да няма никакви симптоми) се оценява на 50%.
В случай на Х-свързано наследяване (когато мутиралият ген е в Х хромозомата на жената), съществува 50% риск майката да предаде гена на сина си, който ще развие ХМТ. Заболяването може да не се появи при раждането на момиче, но синовете (внуците) на дъщерята могат да наследят дефектния ген и заболяването да се развие.
Патогенеза
При всеки вид болест на Шарко-Мари-Тут, нейната патогенеза се причинява от наследствена аномалия на периферните нерви: двигателни (двигателни) и сензорни (чувствителни).
Ако типът CMT е демиелинизиращ, тогава разрушаването или дефектът на миелиновата обвивка, която защитава аксоните на периферните нерви, води до забавяне на предаването на нервните импулси в периферната нервна система - между мозъка, мускулите и сетивните органи.
При аксоналния тип на заболяването самите аксони са засегнати, което се отразява негативно на силата на нервните сигнали, която е недостатъчна за пълноценно стимулиране на мускулите и сетивните органи.
Прочетете също:
Как се предава синдромът на Шарко-Мари-Тут? Дефектните гени могат да се унаследяват по автозомно доминантен или автозомно рецесивен начин.
Най-често срещаният тип, автозомно доминантното унаследяване, се проявява, когато има едно копие на мутиралия ген (носител на един от родителите). Вероятността за предаване на CMT на всяко родено дете се оценява на 50%. [ 4 ]
При автозомно-рецесивно наследяване са необходими две копия на дефектния ген (по едно от всеки родител, който не показва никакви признаци на заболяването), за да се развие заболяването.
В 40-50% от случаите се наблюдава автозомно доминантна наследствена демиелинизация, т.е. CMT тип 1; в 12-26% от случаите - аксонална CMT, т.е. тип 2. А в 10-15% от случаите се наблюдава Х-свързано унаследяване. [ 5 ]
Симптоми Болест на Шарко-Мари-Тоот
Обикновено първите признаци на това заболяване започват да се появяват в детството и юношеството и постепенно се развиват през целия живот, въпреки че синдромът може да се прояви по-късно. Комбинацията от симптоми е променлива и скоростта на прогресиране на заболяването, както и неговата тежест, е невъзможно да се предвиди.
Типичните симптоми на началния етап включват повишена обща умора; намален тонус (слабост) на мускулите на стъпалата, глезените и пищялите; липса на рефлекси. Това затруднява движението на краката и води до дисбазия (нарушение на походката) под формата на по-високо повдигане на краката, често с чести спъвания и падания. Признаците на болестта на Шарко-Мари-Тут при малко дете могат да включват изразена тромавост и нехарактерни за възрастта трудности при ходене, свързани с двустранно увисване на стъпалото. Характерни са и деформации на стъпалата: висок свод (вдлъбнато стъпало) или тежки плоски стъпала, извити (чуковидни) пръсти.
В случай на ходене на пръсти на фона на мускулна хипотония, неврологът може да подозира, че детето има CMT тип 4, при която децата може да не могат да ходят до юношеска възраст.
С напредването на заболяването, мускулната атрофия и слабост се разпространяват към горните крайници, което затруднява фината моторика и извършването на нормални задачи с ръце. Намалените тактилни усещания и способността за усещане за топлина и студ, както и изтръпването на краката и ръцете, показват увреждане на аксоните на сетивните нерви.
При болестта на Шарко-Мари-Тут тип 3 и 6, която се проявява в детска възраст, се наблюдават сензорна атаксия (нарушена координация на движенията и равновесие), мускулни потрепвания и тремор, увреждане на лицевия нерв, атрофия на зрителния нерв с нистагъм и загуба на слуха.
В по-късните етапи може да се наблюдава неконтролируемо тремор и чести мускулни крампи; проблеми с движението могат да доведат до развитие на болка: мускулна, ставна, невропатична.
Усложнения и последствия
Болестта на Шарко-Мари-Тут може да има усложнения и последици като:
- по-чести навяхвания и фрактури;
- контрактури, свързани със скъсяване на периартикуларните мускули и сухожилия;
- сколиоза (изкривяване на гръбначния стълб);
- проблеми с дишането – когато нервните влакна, които инервират мускулите на диафрагмата, са увредени:
- загуба на способността за самостоятелно движение.
Диагностика Болест на Шарко-Мари-Тоот
Диагнозата включва клиничен преглед, анамнеза (включително фамилна), неврологичен и системен преглед.
Извършват се тестове за проверка на обхвата на движение, чувствителността и сухожилните рефлекси. Нервната проводимост може да се оцени с помощта на инструментална диагностика – електромиография или електроневромиография. Може да се изискват и ултразвук или ЯМР. [ 6 ]
Генетичните или ДНК тестове за откриване на най-често срещаните генетични мутации, причиняващи CMT в кръвна проба, са ограничени, тъй като в момента не са налични ДНК тестове за всички видове заболяване. За повече информация вижте Генетично тестване.
В някои случаи се извършва биопсия на периферния нерв (обикновено суралния нерв).
Диференциална диагноза
Диференциалната диагноза включва други периферни невропатии, мускулна дистрофия на Дюшен, миелопатични и миастенични синдроми, диабетна невропатия, миелопатии при множествена и амиотрофична латерална склероза, синдром на Гилен-Баре, травма на перонеалния нерв и неговата атрофия (включително при прищипване между лумбалните дискове на гръбначния стълб), увреждане на малкия мозък или таламуса, както и странични ефекти от химиотерапията (по време на лечение с цитостатици като винкристин или паклитаксел). [ 7 ]
Към кого да се свържете?
Лечение Болест на Шарко-Мари-Тоот
Днес лечението на това наследствено заболяване включва физиотерапия (насочена към укрепване и разтягане на мускулите); трудова терапия (която помага на пациенти с мускулна слабост в ръцете); и използването на ортопедични устройства за улесняване на ходенето. Ако е необходимо, се предписват болкоуспокояващи или антиконвулсанти. [ 8 ]
В случаи на тежки плоскостъпия може да се извърши остеотомия, а при деформация на петата е показана хирургична корекция – артродеза. [ 9 ]
Провеждат се изследвания както на генетичния компонент на заболяването, така и на методите за неговото лечение. Използването на стволови клетки, някои хормони, лецитин или аскорбинова киселина все още не е дало положителни резултати.
Но благодарение на последните изследвания, в близко бъдеще наистина може да се появи нещо ново в лечението на болестта на Шарко-Мари-Тут. Така от 2014 г. френската компания Pharnext разработва, а от средата на 2019 г. текат клинични изпитвания на лекарството PXT3003 за лечение на CMT тип 1 при възрастни, което потиска повишената експресия на гена PMP22, подобрява миелинизацията на периферните нерви и облекчава невромускулните симптоми.
Специалисти от медицинската компания Sarepta Therapeutics (САЩ) работят по създаването на генна терапия за болестта на Шарко-Мари-Тут тип 1. Тази терапия ще използва безвреден аденоасоцииран вирус (AAV) от рода Dependovirus с линеен едноверижен ДНК геном, който ще пренесе в тялото гена NTF3, кодиращ протеина невротрофин-3 (NT-3), необходим за функционирането на нервните клетки на Шван.
Helixmith ще започне клинични изпитвания на разработената в Южна Корея генна терапия Engensis (VM202) до края на 2020 г. за лечение на мускулни симптоми при CMT тип 1. [ 10 ]
Предотвратяване
Превенцията на CMT може да бъде генетично консултиране на бъдещи родители, особено ако някой от двойката има фамилна анамнеза за заболяването. Установени са обаче случаи на de novo точкови генни мутации, тоест при липса на заболяването в фамилната анамнеза.
По време на бременност вероятността от болестта на Шарко-Мари-Тут при бъдещото дете може да се провери чрез биопсия на хорионни въси (от 10 до 13 гестационна седмица), както и чрез анализ на околоплодната течност (на 15-18 гестационна седмица).
Прогноза
Като цяло, прогнозата за различните видове болест на Шарко-Мари-Тут зависи от клиничната тежест, но във всички случаи заболяването прогресира бавно. Много пациенти имат увреждания, въпреки че това не намалява продължителността на живота.